Акромегалия — хроническое заболевание, вызванное гиперпродукцией гормона роста и ИФР-1, сопровождающееся изменением внешности, головными болями, артралгией. Заболевание связывают с нарушением функции гипофиза, среди причин большую часть составляют генетические мутации. В статье представлен обзор клинического случая эндоскопического транссфеноидального удаления аденомы гипофиза у пациента с множественной эндокринной неоплазией 1-го типа (MEN1).
При раннем обнаружении акромегалия поддается лечению, поэтому осведомленность пациентов, имеющих предрасположенность к этому заболеванию, играет принципиальную роль. В это понятие входит не только знание симптомов акромегалии, но и семейный анамнез, поскольку случаи генетических болезней у близких родственников имеют большое значение.
Причины акромегалии
Акромегалия развивается из-за гиперпродукции гормона роста (ГР) и ИФР-1. Повышенный уровень ГР может быть связан как с аденомой гипофиза, так и с гиперплазией ткани гипофиза. В литературе описаны мутации в составе различных генетических заболеваний, приводящих к гиперплазии гипофиза. Почти в половине случаев могут быть обнаружены те или иные генетические мутации по наследственной линии.
Акромегалия может быть одним из синдромов при множественной эндокринной неоплазии 1-го и 4-го типа (МЭН1, 4) или феохромацитоме. Диагноз МЭН1 устанавливается на основании наличия двух и более эндокринных образований (гипофиз, паращитовидная железа, нейроэндокринная опухоль поджелудочной железы) или наличия одной опухоли и подтвержденной мутации МЭН1 у одного ближайшего родственника. Носителям данной мутации необходимо выполнять регулярный скрининг, тогда лечение доброкачественных новообразований можно будет начать на ранних этапах.
Семейная изолированная аденома гипофиза может быть заподозрена у пациента с наличием в семье случаев аденом гипофиза при отсутствии других патологий эндокринной системы. В 10% случаев данная группа пациентов имеет генетическую мутацию AIP белка. Изолированные аденомы с мутацией AIP белка характеризуются ранним дебютом заболевания (до 30 лет), имеют крупные размеры, экстраселлярный рост, высокую частоту апоплексии гипофиза.
Вторым по частоте после AIP ассоциированной аденомы гипофиза является Х-сцепленный акрогигантизм (XLAG). Встречается у 10% пациентов с клиникой гигантизма, чаще у женщин.
Виды акромегалии
По данным литературы, всего описано 40 случаев данной генетической мутации.
Синдром МакКьюна — Олбрайта (MAS) может сопровождаться как гиперплазией, так и аденомой гипофиза, при этом синдром акромегалии встречается в 30% случаев. Средний возраст пациентов составляет 20 лет, чаще мужчины со смешанной секрецией. Специфическими клиническими проявлениями данного синдрома могут быть фиброзная дисплазия, преждевременное половое созревание и кожные изменения по типу «кофе с молоком».
Комплекс Карни включает в себя образование или гиперплазию ткани гипофиза, образование надпочечников. Клинически акромегалия может наблюдаться у 10% пациентов и проявляется к 30 годам.
Акромегалия: клинический случай
В статье описан клинический случай пациента Б. 39 лет, поступившего в нейрохирургическое отделение с жалобами на общую слабость, головную боль, снижение остроты зрения на правый глаз, высокие показатели глюкозы в крови, изменение внешности (огрубление черт лица (акромегалия черепа), увеличение пальцев рук), повышение артериального давления. Изменение внешности было постепенным начиная с 25 лет, однако пациент не придавал этому значения.
Пациент был госпитализирован в стационар, На основании объективных внешних данных была заподозрена акромегалия. По результатам МРТ головного мозга, повышенного уровня СТГ, ИФР-1 диагноз был подтвержден. Консультирован нейрохирургом, было рекомендовано оперативное лечение. При объективном обследовании в неврологическом статусе была зафиксирована правосторонняя гемианапсия.
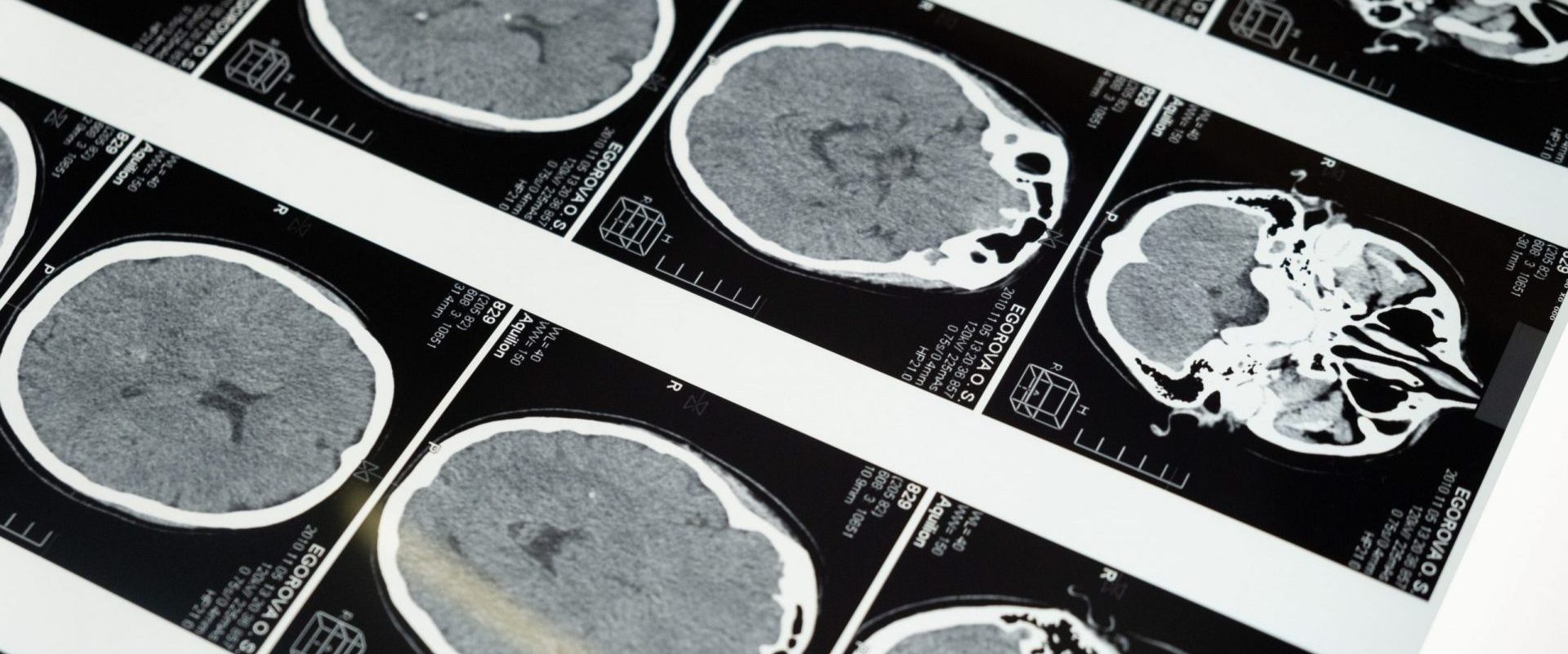
По данным предоперационного обследования на МРТ головного мозга с в\в контрастированием обнаружено объемное образование кистозно-солидной структуры, размерами 25х33х37 мм. Лабораторные данные: ИФР-1 750 нг / мл, СТГ 13,97, пролактин — 3180 мМЕ / мл.
Пациенту путем транссфеноидальной хирургии выполнено субтотальное удаление аденомы гипофиза (выбор оптимальной хирургической тактики в современной нейрохирургии определяется размерами опухоли и степенью ее инвазии в кавернозный синус). Гистологическое заключение: СТГ и ПРЛ секретирующая аденома гипофиза.
Пациент был выписан на третьи сутки после оперативного лечения. Жалоб на снижение остроты зрения, головную боль, осложнений не было. Однако через 3 месяца после оперативного лечения визуализировалась остаточная опухолевая ткань с кистозной перестройкой, размерами 9х11х10 мм. Также сохранялся высокий уровень ИФР-1 – до 620 нг/мл, несмотря на прием лекарств. После смены терапии уровень ИФР-1 снизился до 250 нг/мл.
Пациенту был проведен молекулярно-генетический анализ, который показал патогенный вариант гена MEN1 в гетерозиготном состоянии. Также положительный результат был получен у его сына.
Акромегалия: клинические рекомендации
Обращает на себя внимание длительный анамнез заболевания в виде характерных для акромегалии внешних изменений с 25 лет, на которые пациент не обращал внимания. На момент постановки диагноза пациент имел ряд системных осложнений (гипергликемия, артериальная гипертензия), высокие предоперационные уровни СТГ, ИФР-1, большой размер опухоли и тотальную инвазию опухоли в кавернозный синус. Признаки гиперкальциемии и высокий уровень паратгормона, по данным лабораторных методов обследования, наличие объемного образования паращитовидной железы указывали на наличие у пациента синдрома множественной эндокринной неоплазии МЕН1, которая была подтверждена молекулярно-генетическим исследованием.
Необходимость в генетическом тестировании пациентов с акромегалией возникает при анализе возрастного дебюта заболевания, семейного анамнеза, при наличии других образований эндокринной системы. Повышение осведомленности пациента о причинах своего заболевания, поиск других эндокринных патологий являются важными задачами для развития современной неврологии. Скрининг родственников пациента необходим для ранней диагностики начала заболевания. Лечение акромегалии на ранних стадиях позволяет значимо улучшить исходы хирургического, консервативного лечения пациентов.
Ознакомиться с полной версией материала и другими публикациями вы можете, оформив подписку на журнал «Вестник неврологии, нейрохирургии и психиатрии» ИД «ПАНОРАМА».